
Reading time: 5 minutes
Chris Wang
Like Keanu Reeves dodging bullets within The Matrix, cancer cells can dodge even carefully designed anti-cancer agents. Scientists strive to design “silver bullets,” ones which target cancer cells while sparing healthy normal cells. This “silver bullet” approach is appealing to researchers due to the promise of improved therapeutic response and fewer side effects compared to conventional chemotherapies. While targeted treatments have revolutionized cancers such as chronic myeloid leukemia, turning the disease from a death sentence to a manageable chronic condition, not all cancers have such treatment options. However, this situation is changing as researchers are constantly finding new targeted treatments, including one called BRAF inhibitors. These drugs are particularly effective in killing cancers that utilize the BRAF pathway to multiply, including the most lethal type of skin cancer: melanoma.
Up to 50% of metastatic melanomas express a mutated form of the BRAF protein that is always active, allowing the melanoma cell to grow uncontrollably. BRAF activity is normally regulated and can be turned off and on when needed, but when BRAF becomes mutated, its activity is always on and the cell continuously divides. This motivated researchers to find drugs that could inhibit signaling through mutant BRAF and impair the growth of melanoma cells (see Figure 1). There are now three FDA approved BRAF inhibitors (vemurafenib, encorafenib, and dabrafenib) which have been shown to increase overall survival time from a median of 8 months on traditional chemotherapy to around 12 to 16 months. However, this response is often temporary; many patients whose tumors initially respond to BRAF inhibitors eventually develop resistance, and their disease progresses despite treatment.
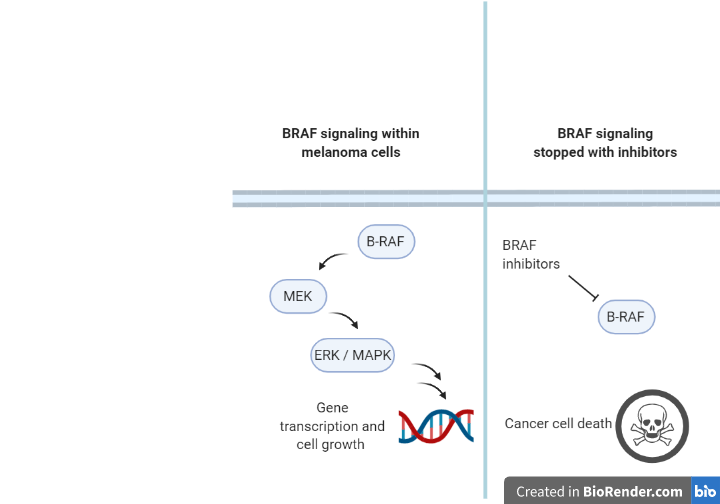
Melanoma cells, like many cancer cells, are constantly changing and can gain mutations (shown in Figure 2) that restore the activity of key growth signaling pathways, some of which are BRAF dependent or independent. By restoring these signaling pathways, melanoma cells can re-enable uncontrolled cell division, even in the presence of BRAF inhibitors. For example, amplification of the BRAF gene results in an excess of BRAF protein, so that the limited amount of BRAF inhibitors a patient is dosed with cannot fully block signaling. Truncating the BRAF gene creates a BRAF protein that does not bind inhibitors but can still activate MEK. Melanomas can also overcome BRAF inhibition and maintain growth signaling through adaptations that are independent of BRAF itself; for example activating a BRAF relative called CRAF that can also activate MEK, activating non-MEK proteins that promote ERK activity, or event co-opting non-BRAF dependent growth pathways such as the PI3K pathway. It is important to note that the mutations outlined in Figure 2 are both simplified and not all-encompassing. Additionally, BRAF inhibitor-resistant melanoma likely harbors multiple mutations conferring resistance, rather than just one.
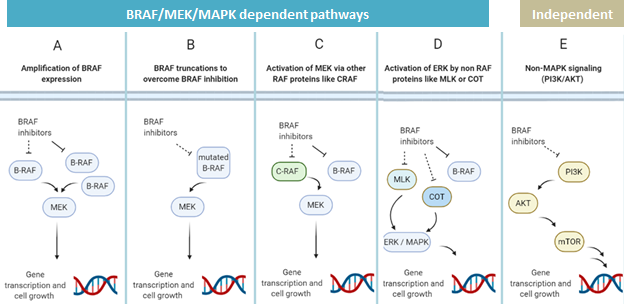
This phenomenon spurred researchers to develop treatments that overcome BRAF resistance through the alternative growth signaling pathways mentioned in Figure 2. One of their therapeutic targets included the protein MEK, which is activated downstream of BRAF. With the idea that two “bullets” would be harder to dodge than one, scientists hypothesized that BRAF inhibitor resistant patients might respond to treatment with BRAF and MEK inhibitors at the same time. This hypothesis was finally right: trials showed dual inhibition improved the median overall survival of metastatic melanoma patients from less than 18 months on BRAF inhibition alone to around 22 to 26 months. This led three BRAF/MEK inhibitor combinations (vemurafenib + cobimetinib, encorafenib + binimetinib, and dabrafenib + trametinib) to be recommended as first line treatment options for BRAF mutant metastatic melanoma.
To better understand the concept behind the mechanism of dual BRAF and MEK inhibition, see Figure 3. While pathways A-C (Figure 3) are resistant to BRAF inhibition alone, simultaneous dual inhibition of BRAF and MEK allows for the blocking of the BRAF-pathway despite BRAF amplification, truncation, or BRAF-independent MEK activation through stopping the pathway at a more downstream site. However, even as dual inhibition demonstrated improved survival compared to either BRAF or MEK inhibition alone, eventually many patients who received dual therapy also progressed despite treatment. This resistance could be partially explained by the fact that dual BRAF and MEK inhibition disrupts only some of the ways melanoma cells can resume growth pathways (Figure 3 panels A through C) shown while leaving others intact, shown in (Figure 3 panels D and E). Thus, there remains a need for effective treatments against dual BRAF/MEK inhibitor resistant melanomas, and current research is exploring inhibition of downstream effectors in the MAPK or PI3K pathways towards this end.
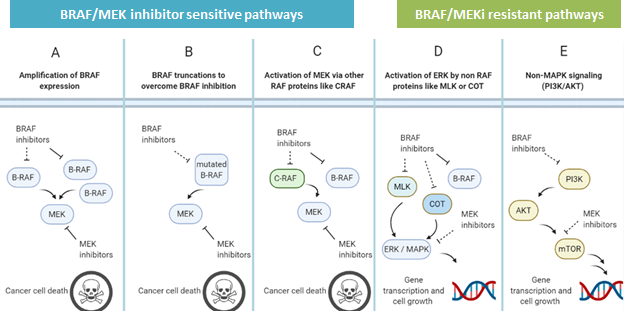
While there still lies a long road ahead before a majority of BRAF mutated melanoma patients have lifelong survival without treatment resistance, someday scientists hope to develop a new “silver bullet” that even BRAF/MEK inhibitor resistant melanoma cannot dodge. And the tale of how researchers discovered the mechanisms of drug resistance in melanoma to successfully create targeted therapies signals our progress towards that goal.
Edited by Emily Costa
Abbreviations used: B-Rapidly Accelerated Fibrosarcoma, B-RAF; FDA, Food & Drug Administration, FDA; Mitogen-Activated Protein Kinase Kinase, MEK; Extracellular signal-Regulated Kinase/Mitogen-Activated Protein Kinase, ERK/MAPK; C-Rapidly Accelerated Fibrosarcoma, C-RAF; Mixed Lineage Kinases, MLK; Cancer Osaka Thyroid oncogene, COT; Phosphoinositide 3-Kinase, PI3K; Protein kinase B, AKT; Mammalian Target of Rapamycin, mTOR.
For more information about BRAF and its role in cancer; check out these past OncoBites articles:
For more information about how cancer cells develop resistance to treatment; checkout these past OncoBites articles:
Reference
Sanchez JN, Wang T, Cohen MS. BRAF and MEK Inhibitors: Use and Resistance in BRAF-Mutated Cancers. Drugs. 2018 Apr; 78(5): 549–566.
Image
Image from: Jones, Ralph. “Inside Keanu Reeves’s bullet time scene: how The Matrix changed cinema forever.” The Telegraph, August 29th, 2019. https://i.kym-cdn.com/entries/icons/original/000/018/083/image.jpg. Accessed August 9th, 2020.

{kind=link}
Really interesting, thanks for sharing this!
As an incoming cancer research PhD candidate, I’m so happy to have come across your site! Exactly what I was looking for 😀
LikeLike