Reading time: 4 minutes
Suchitra Mitra
Normal cells grow, age, and ultimately die to be replaced by new cells. Cancer cells, on the other hand, experience cell cycle malfunctions and grow out of control to become invasive, spreading undesirably and harmfully.
This anomalous cell proliferation is a consequence of genetic alterations, or changes in the basic units of heredity. One of the main types of genes affected by cancer-causing genetic alterations are proto-oncogenes. Once altered, they form oncogenes that overexpress, leading to cancer.
c-Myc is one of the most common oncogenes that gets upregulated in ~70 % of human cancers.
For example, in almost all cases of Burkitt’s lymphoma, chromosomal translocations (an abnormality where a portion of chromosome breaks and attaches with another chromosome) activate c-Myc. In addition, 20-30% of breast and prostate cancers result from c-Myc amplification, and 70-80% of colon cancers result from de-regulation of c-Myc’s transcription control. Hence, c-Myc is often referred to as the ‘oncogene from hell,’ and its inhibition has been a traditional approach in cancer therapy.

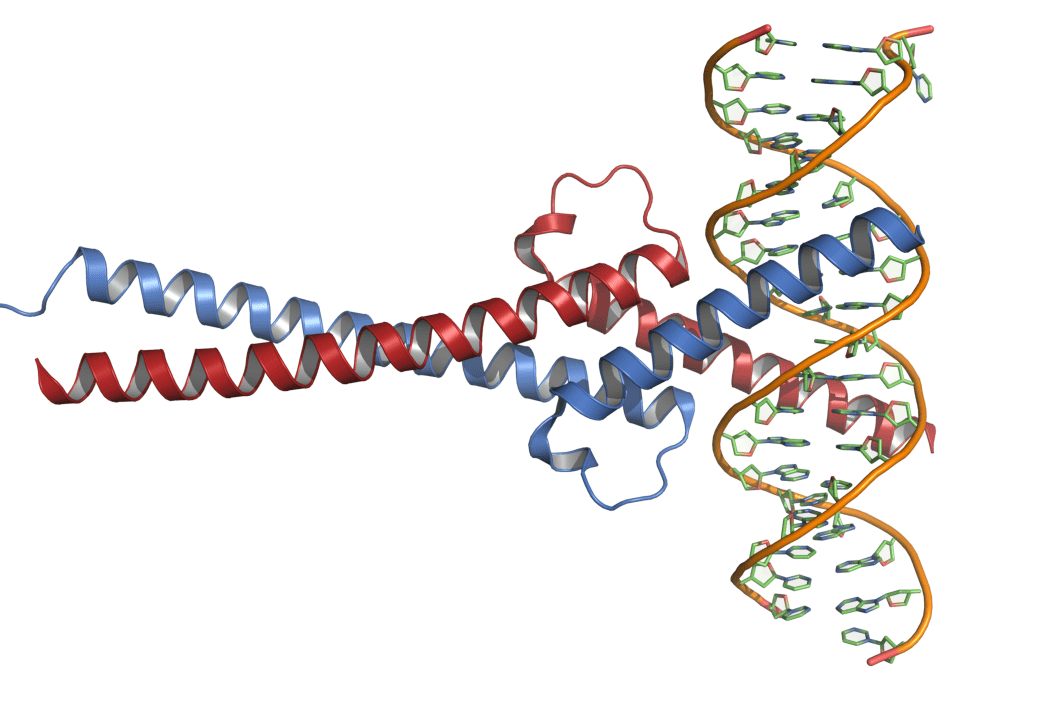
As a proto-oncogene, c-Myc plays an important role in cell growth, differentiation, metabolism, apoptosis, and many other processes (Figure 1). The c-Myc protein or the Myc protein (c- dropped for simplicity) has a specific secondary structure that only allows it to bind with a protein having a similar structure, its partner, Max. The Myc-Max complex is highly stable because of protein-protein interactions, which involve a combination of ionic bonding, H-bonding, and hydrophobic packing. Myc and Max are perfect partners, as Max serves as Myc’s positive regulator and is essential to Myc’s functions, including DNA binding and transcription. Myc doesn’t self-assemble and remains unstructured until partnered with Max. Myc and Max together adopt an alpha helical structure that anchors them to the DNA (Figure 2). The Myc-Max complex binds to the promoter region of DNA for a large section of genes. They code for a major portion of genome that influences cellular processes to a large extent, involving DNA replication, transcription, mitochondrial and ribosomal biogenesis, cell cycle, and so on. Myc-Max also stimulates RNA polymerases I, II, and III that further stimulate transcription of more genes. Under normal physiological conditions, Myc is tightly regulated by various cellular checkpoints and has a short half-life of ~20 minutes. However, in cancer, this homeostasis is disturbed, and Myc becomes free of any checkpoints or regulations – leading to overexpression, which eventually leads more and more cells to grow, causing cancer.

Although rarely mutated, Myc is frequently upregulated in almost all human cancers. However, there are no drugs yet approved that target Myc. This is because the structure of Myc lacks a specific drug binding pocket. The location of Myc is also elusive – Myc being a nuclear protein is difficult to target via drugs. Moreover, the role of Myc in metastasis is controversial with some reports stating that Myc inhibition could induce metastasis. These factors almost make Myc undruggable.

In spite of these challenges, both direct and indirect inhibition strategies of Myc are reported in the literature. One of the critical anti-Myc therapies uses small molecule and miniprotein/peptide inhibitors. These drugs consist of lower molecular weight compounds that can be easily synthesized and intuitively designed to reverse the protein-protein interaction either involving directly the Myc-Max complex or indirectly with other cellular proteins affecting Myc (Scheme 1). However, small molecule drugs may induce significant toxicity in vivo, and they must be fine-tuned for maximum potency. Lack of target specificity and bioavailability are other critical challenges involved with small molecule drugs. Mini-proteins or peptides can be more potent in vivo.
Some of the indirect approaches involve targeting cellular proteins responsible for Myc transcription, translation, or stability. One such example is targeting the bromodomain containing 4 protein (BRD4), which regulates Myc transcription. BRD4 recruits a positive transcription elongation factor, which helps RNA polymerase II in transcription elongation. JQ1 is a small molecule inhibitor that inhibits BRD4 binding to Myc’s DNA sequences that bind to transcription factors for enhanced transcription. GSK525762 is another similar BRD4 inhibitor, which is currently in the early phases of clinical trials for immune cell cancers and solid tumors (ClinicalTrials.gov: NCT01943851, NCT03266159).
Direct approaches to Myc inhibition mainly involve inhibiting Myc’s stability or disrupting the Myc-Max complex and Myc’s binding to DNA. Without Max, Myc would fail to bind to DNA, and hence its proliferative activity can be inhibited. One example is the 90 amino acid mini-protein, Myc mutant – Omomyc. It has a similar structure to Myc with four mutations, which alter the protein-protein interaction, and consequently, it can dimerize with Myc, Max, and itself. Hence, it can trap Myc, disrupt the Myc-Max complex, and also occupy DNA with inactive dimers (Max-Ommomyc, Omomyc-Omomyc, Myc-Omomyc). Omomyc is one of the most studied protein inhibitors of Myc and is expected to reach clinical trials soon.
These direct and indirect approaches to target Myc give us hope for more selective and potent clinically available drugs in the future.
Edited by Isabel Newsome
References.
- Chen, H., Liu, H. & Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Sig Transduct Target Ther 3, 5 (2018).https://doi.org/10.1038/s41392-018-0008-7
- Massó-Vallés D, Soucek L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells. 2020, 9, 883. doi: 10.3390/cells9040883.
Header image: Crystal structure of Myc and Max in complex with DNA.

{kind=link}