Reading time: 5 minutes
Keighley Reisenauer
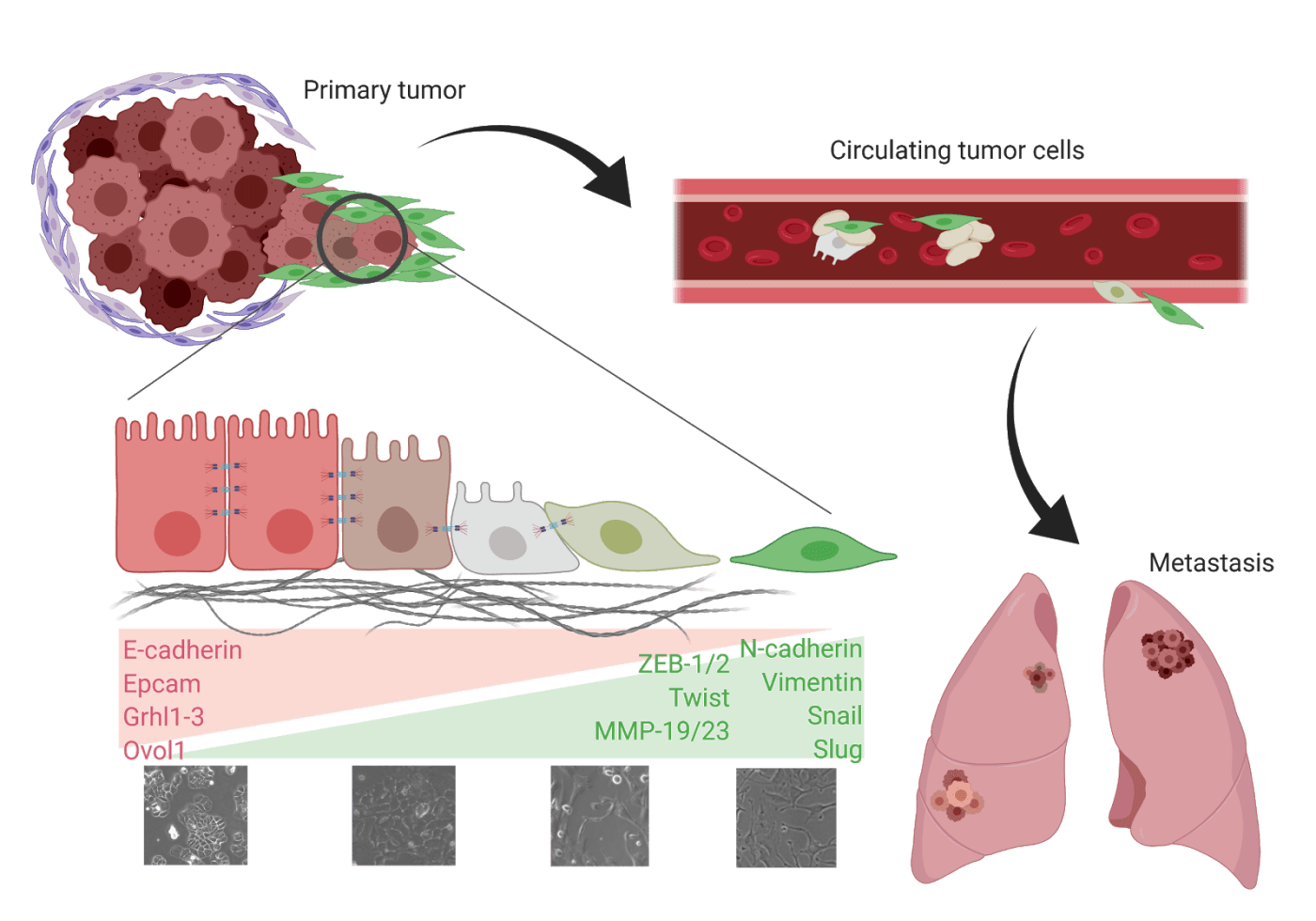
Google “EMT,” and you’re likely to see images of paramedics and ambulances. However, there is another EMT that plays a much different role in disease and illness. Epithelial to mesenchymal transition (EMT) is normally associated with processes such as wound healing and development. Cancer hijacks EMT to participate in the separation of the tightly-bound cells of epithelial tumors (breast, lung, ovarian, etc.) from the main tumor and begin the metastatic cascade. This process involves a temporal activation of mesenchymal-associated genes and downregulation of epithelial genes. The cells lose cell-to-cell adhesion and apical-basal polarity, become anoikis resistant, and – worst of all – become likely to metastasize and evade chemotherapy. This last point is what has left researchers baffled at best and in stark opposition at worst since cancer-related EMT was first described.
EMT was first characterized into three types during a 2007 meeting on EMT in Poland and a subsequent meeting in March 2008 at Cold Spring Harbor Laboratories. Specifically, Type III EMT is associated with carcinomas. While thousands of papers have been published on the topic of this type III EMT, patients still face related therapeutic challenges such as disease progression, recurrence, and therapy resistance. Understanding what drives EMT in cancer, how it is activated, and the results of its activation are the only way to alleviate these outcomes for patients and promote improved prognosis.
To understand EMT’s role in metastasis and the surrounding controversy, look no further than a series of papers that nearly turned the field upside down. EMT was the leading explanation for how and why metastasis happens. The idea began with studies that showed how epithelial tumor cells that had detached from the main tumor would die due to lack of cell-to-cell adhesion or immune attack. It was only by losing those traits and acquiring mesenchymal characteristics that tumor cells could enter, survive through, and exit the circulation. This idea was not 100% universal, though, with papers carving out caveats and stipulations that narrowed and narrowed the applicability of this concept. One such paper was published in 2013 and describes that tumor cells found in the circulation are not the homogenous mesenchymal phenotype described in the EMT-metastasis hypothesis. Rather, the authors described the presence of heterogeneous, small clusters of circulating tumor cells in the blood, which opened the door to the possibility that cells could metastasize without EMT. This paper opened the door to further criticisms and the most controversial paper to date was released by Fisher et. al. in Nature in 2015. In it, authors challenged the leading consensus that EMT was required for cancer metastasis by studying which types of cells were actually seeding metastatic sites. Using a mouse lineage-tracing model, these researchers created a genetic system that would turn all epithelial cancer cells red by expressing red fluorescence protein (RFP) under a Cre-Lox reporter system. This system allowed for cells activating EMT to then turn permanently green by expressing green fluorescence protein (GFP). Researchers demonstrated the effectiveness of this system by showing that the primary tumors were predominantly RFP-positive, but then also that the metastases were also mostly RFP-positive, suggesting that these cells did not activate EMT in order to metastasize. However, the paper does concede that, following chemotherapy, cells activate EMT to colonize metastatic sites.
Most interestingly, though, was the rebuttal paper that followed the 2015 Fisher paper. This paper came out of Dr. Robert Weinberg’s lab, a scientist known as the “Father of EMT.” He has dedicated his career to the notion that EMT drives metastasis, so it was almost expected from the scientific community that his rebuttal was imminent. By July of the following year, the Brief Communications Arising article was released from the Weinberg lab on the premise that the Fisher paper did not, in fact, prove EMT did not happen. The rebuttal leaned on the decades of research describing EMT as a complex process happening along a network of signaling channels and “related cell-biological programs.” The 2016 Weinberg article explored the inefficiency of the mouse model used in the Fisher paper, stating that some of the cells may not have expressed the Cre-Lox reporter system and some of the cells may not have appropriately activated GFP expression, indicating a leaky system. Secondly, they challenged the EMT marker that the Fisher paper relied heavily on, stating that Fsp1 is not the “critical gatekeeper gene of EMT initiation” as Fisher et. al. described it. To strengthen this, the rebuttal article yields that EMT-research has described it as an imperfect system, where even some of the key proteins, such as vimentin, are not always expressed. When the Weinberg lab used the same system as Fisher, but used a protein more tightly associated with EMT for the knockout, they noted that metastases nearly disappeared with this loss of ability to activate EMT. Ultimately, the rebuttal paper “conclude[d] that the Fischer et. al. report was insufficient to rule out the contribution of EMT to metastasis.”
This dynamic interplay of research goes to show the complexity of EMT and its intricate role in metastasis. Other major papers that have linked EMT to characteristics of worsened prognosis are a 2008 paper demonstrating that EMT generates cells with stem cell-like properties, implicating EMT further in chemoresistance and disease recurrence, and a 2017 paper exploring the sensitivity of EMT-activated cells to immune therapies on the basis that the EMT signature correlates with tumor types that respond best to certain immune system-based treatments and with tumors that have increased expression of multiple immune checkpoint markers. Though this field is evolving rapidly and EMT-related research is pouring out at incredible rates, a 2019 review says it best: “[scientists] are still far from reaching a complete understanding of the biological mechanism and pathological contribution of epithelial-mesenchymal plasticity in cancer.” The most promising studies are utilizing advanced techniques and technologies such as lineage tracing animal models, patient derived xenografts, high-resolution intravital imaging, and single-cell sequencing.
So, while EMT may not be the first thing discussed when evaluating cancer treatment plans or revolutionary therapeutic breakthroughs, understanding it more fully will be the foundation needed to bolster cancer research and treatment options for patients fighting this complex disease.
Edited by Zoey Tang
Works discussed:
Boyer, B., Vallés, A.M., and Edme, N. (2000). Induction and regulation of epithelial-mesenchymal transitions. Biochem. Pharmacol. 60, 1091–1099
Lu, W., and Kang, Y. (2019). Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Developmental Cell 49, 361–374.
Fischer, K.R., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S.T.C., Choi, H., El Rayes, T., Ryu, S., Troeger, J., et al. (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476.
Aceto, N., Toner, M., Maheswaran, S., and Haber, D.A. (2015). En Route to Metastasis: Circulating Tumor Cell Clusters and Epithelial-to-Mesenchymal Transition. Trends in Cancer 1, 44–52.
Ye, X., Brabletz, T., Kang, Y., Longmore, G.D., Nieto, M.A., Stanger, B.Z., Yang, J., and Weinberg, R.A. (2017). Upholding a role for EMT in breast cancer metastasis. Nature 547, E1–E3.
Mani, S.A., Guo, W., Liao, M.-J., Eaton, E.Ng., Ayyanan, A., Zhou, A.Y., Brooks, M., Reinhard, F., Zhang, C.C., Shipitsin, M., et al. (2008). The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 133, 704–715.
Noman, M.Z., Janji, B., Abdou, A., Hasmim, M., Terry, S., Tan, T.Z., Mami-Chouaib, F., Thiery, J.P., and Chouaib, S. (2017). The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 6, e1263412.
