
Reading time: 7 minutes
Felicites Rapon
Hepatitis C virus (HCV) is the causative agent of hepatitis C, an infectious disease of the liver causing inflammation and necrosis. HCV infection can be either acute (which can be cleared without any medicines) or chronic. The latter can be due to environmental factors such as alcohol usage but also due to host factors such as gender, age or genetics, which can lead to cirrhosis. Cirrhosis is characterized by fibrosis (scar tissue) and regenerative nodules. Once cirrhosis occurs, patients are more at risk of a liver cancer emergence called hepatocarcinogenesis, resulting in hepatocellular carcinoma (HCC), a primary liver cancer.
HCV is therefore known as an oncogenic virus, as it can ultimately lead to cancer. The World Health Organization (WHO) has estimated that 58 millions people have chronic hepatitis C virus infection and in 2019 around 290 000 died from hepatitis C, mostly from cirrhosis or HCC [1]. In addition, liver cancer is the 6th most frequent cancer in the world (5th in men, 9th in women) [2]. Though antiviral therapy exists for HCV, it is insufficient due to various side effects and also the development of drug resistance by the virus. Drug resistance occurs due to the virus changing its genome through mutations which will bypass and resist antiviral drug mechanisms. Therefore, it is crucial to develop vaccines as, to date, none has been effective against HCV.
This is where the story of the multi-epitope vaccine candidate against hepatitis C virus [3] begins. We live in a world where current technologies such as our smartphone can easily beat the NASA 1960s computers that sent men to the moon. In the field of biology, numerous computing tools have been developed in the last 20 years, we call them bioinformatics tools. They allow biologists and more generally scientists to save time – going faster on certain experiments but also make savings as these tools can sometimes replace some costly experiments. Recently, an Iranian team [3] of scientists have been able to build, using only computational methods (also known as in silico), new types of vaccines against HCV.
These vaccines were made possible due to a new field of science that uses these computational tools, called reverse vaccinology. This is based on the selection of the best components for the creation of a vaccine. These components, called antigens, are essential and have to be chosen carefully to ensure an appropriate immune response. This method has previously been fruitful as in 2015 the FDA approved Bexsero [4] which is a vaccine against serogroup B Meningococcal disease which was also first designed in silico. What was interesting in this construction was the fact the vaccine is composed of several epitopes also called multi-epitopes allowing a better and more efficient immune response compared to a single one.
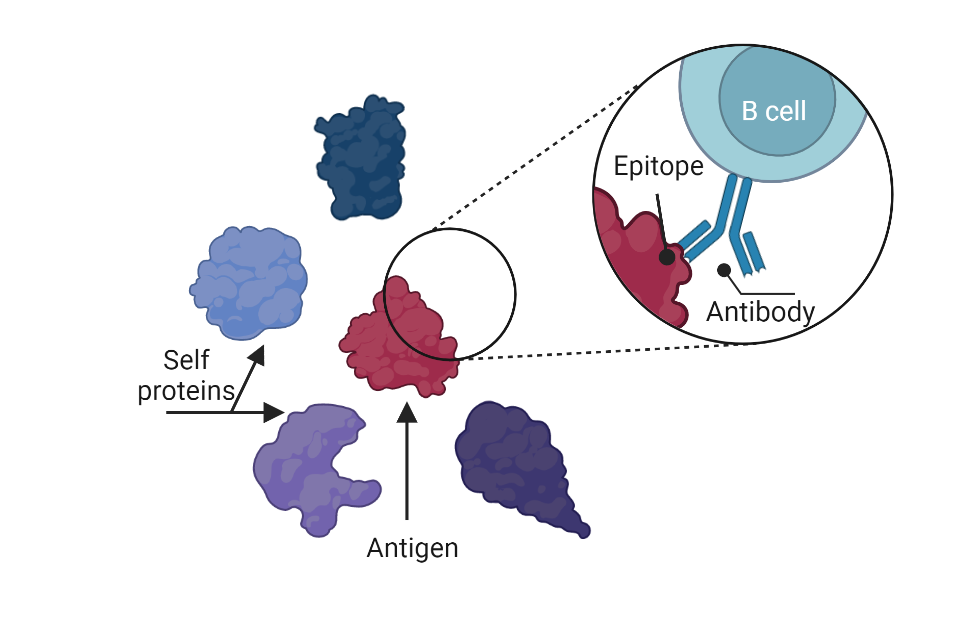
Figure 1 : B cell receptor recognizing an epitope, part of an antigen (non-self protein in red). Image created with BioRender.com
An epitope, in immunology, is a small part of an antigen which is itself a protein detected by the human body as a foreign protein (non-self). This epitope, once bound to a host (from the human body) immune cell via receptors on its surface, will lead to an immune response (Figure 1).
In the present study, the designed vaccine protects against HCV. So, how did this team build a multi-epitope vaccine against HCV? The flow chart below shows how the scientists created a vaccine with only bioinformatics tools. Behind these obscure words is hidden a very simple and solid plan to produce efficiently in silico any multi-epitope vaccines.
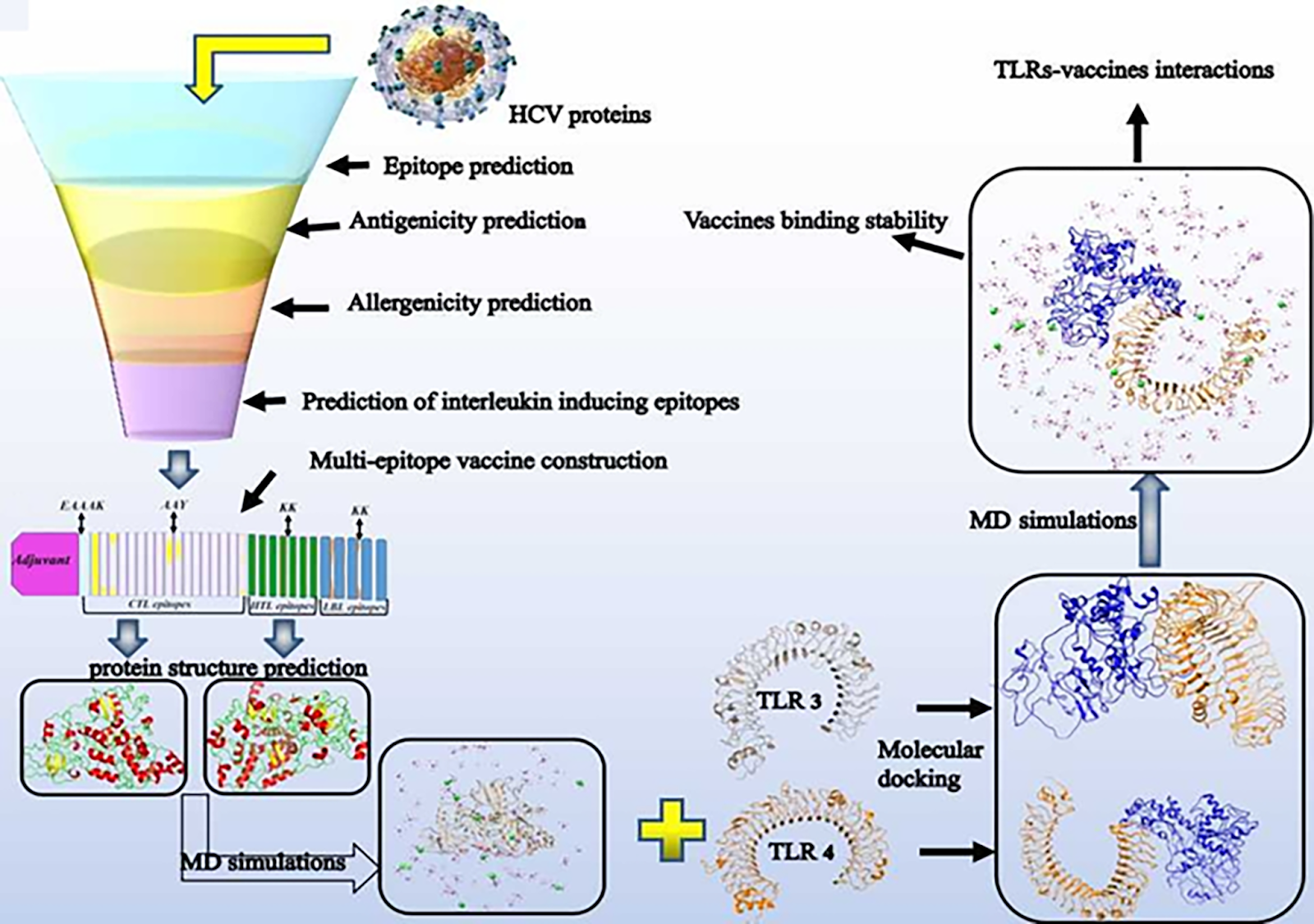
Figure 2 : Flowchart describing chronologically the different steps used by the scientists to design the candidate vaccine. [3]
If we follow this flow chart, first, researchers were considering what sort of vaccine to create. To date, treatments against HCV are not as effective as they could be, especially as there is an underlying problem related to drug resistance as mentioned above. This resistance manifests through mutations on viruses to evade the immune system.
HCV proteins and epitope prediction:
To circumvent this problem they decided to produce a vaccine based on epitopes. Those selected virus proteins no matter their positions on the virus (structural or non-structural proteins) will increase the diversity of epitopes and allow a more specific immune response.
Moreover, as there are different virus strains that are several versions of the same virus with different mutations, the selected proteins will be chosen where there are few mutations. That way, the vaccine should be able to kill different strains of the virus.
Antigenicity and allergenicity prediction:
After determining the epitopes, the researchers had to predict their reaction with the immune system especially with adaptive immune cells. Indeed, the adaptive immune system plays a crucial role for vaccines as this system is based on the memory of cells to recognize infectious molecules (such as antigens) but also on an effective immunization (response of the body or host cells) against those molecules. In this way, binding affinity assays have been carried out using online software, between different immune cells such as cytotoxic T lymphocytes or helper T lymphocytes or even B lymphocytes with epitopes.
The aim of binding affinity assays is to measure the strength of binding (affinity) between two molecules: the ligand (in this example epitopes) and the target biomolecule ( immune cells that will trigger the adaptive immunity which is essential for the vaccine efficacy).
The two molecules do not directly bind, but rather go through molecules called MHC (Major Histocompatibility Complex) which will present epitopes to the adaptive immune cells. The assays used by the team showed a very good affinity. It was especially important with B cells as they will produce antibodies which recognize HCV antigens. Predicting the fact that interleukins have been produced after the binding of an epitope with an immune cell is also a key point as they play a role of messenger among the different immune cells.
Estimates about the possible toxicity or allergenicity of epitopes have been realized with bioinformatics tools to remove the ones that could lead to allergic or even toxic reactions with the body that are not wanted for the creation of vaccines.
Multi-epitope vaccine construction:
With the well-fitted epitopes (in that they have been selected as the best among several), the researchers create the final vaccine construct. They link the different epitopes with linkers and add some adjuvant (to help in Latin) molecules to improve immunogenicity properties of the vaccine (the adjuvants increase the efficacy of the immune response). The two adjuvants used (human β-defensin 2 and 50S ribosomal protein L7/L12) in the study are agonists of receptors of innate immunity (Toll-like receptor, TLR), they will be able to activate TLR receptors as if they were molecules of the virus. Indeed, TLR recognizes antigens and will produce an immune response.
From this construct, the researchers then predict the conformation in 3D of the final protein based on the sequence of amino acids. With online bioinformatics software, they have been able to obtain predictions about the 3D conformation.
MD simulations:
Molecular dynamics (MD) simulations have been conducted on computers to analyze the stability over time of the predicted 3D conformation of the vaccine by measuring parameters such as RMSD (Root mean square deviation) that allow to measure, for example, distances between two sets of atoms of a protein over the time. The values obtained as a function of time will describe a trajectory showing us how much the protein shape has changed over a period of time.
Molecular docking:
After the simulations, the next question was to know whether or not the predicted 3D conformation of the vaccine could match with the main 2 types of receptors called TLR (for Toll Like Receptors) on certain immune cells. For that, molecular docking with informatics tools have been carried out. It allows them to see which amino acids or at least which regions of the vaccine were bound to the receptors. Once assembled, MD simulations found that the TLR-vaccine combination was stable over time.
In the end, thanks to bioinformatics, it seems simple and fast to create multi-epitope vaccines. These vaccines present numerous advantages as they have been designed to lead a very efficient immune response. We realize it is easy to standardize this vaccine on a large scale at low cost. It is also easier for a team to work with bioinformatics tools as they don’t manipulate infectious molecules. However, we have to keep in mind that this team has only worked, in silico, with computers and further studies on this specific vaccine will need to be carried out by other teams working this time in vitro on cells or in vivo with different animal models and finally on the human body with clinical trials. If such a vaccine candidate could be found thanks to this process, it could not only reduce virus load at the very beginning of the infection but perhaps eradicate the virus completely and in a sustainable way through adaptive immunity memory. This can only be a good thing for reducing the burden of cirrhosis and eventual liver cancers.
Edited by Sushma Teegala
References
[1] World Health Organization. “Hepatitis C.” June 24 2022. https://www.who.int/news-room/fact-sheets/detail/hepatitis-c
[2] World Cancer Research Fund International. “Liver cancer.” https://www.wcrf.org/diet-activity-and-cancer/cancer-types/liver-cancer/
[3] Behmard E et al, “Design of a novel multi-epitope vaccine candidate against hepatitis C virus using structural and nonstructural proteins: An immunoinformatics approach”, PLOS ONE, 2022
[4] Novartis, “Novartis Bexsero® vaccine approved by FDA for the prevention of meningitis B, a leading cause of bacterial meningitis in the US.” January 23, 2015. https://www.novartis.com/news/media-releases/novartis-bexsero-vaccine-approved-fda-prevention-meningitis-b-leading-cause-bacterial-meningitis-us
Header image: https://commons.wikimedia.org/wiki/File:Hepatitis_C.jpg

{kind=link}
Leave a comment