Reading time: 5 minutes
Anamika Bandyopadhyay, PhD
Chemotherapy transformed cancer treatments in the 20th century, but its lack of specificity remained a major limitation throughout. As time progressed, hormone therapy, immunotherapy and other targeted therapies were developed. In recent times, cell-penetrating peptides (CPPs) have been used to deliver otherwise impermeable drugs directly into cells. However, here too, specificity poses a challenge. The cell-penetrating peptides require a positive charge (protonation) to interact with the negatively charged cell membrane lipids to gain entry to the cell. These CPPs are typically composed of arginine and lysine residues. Both are positively charged at physiological pH. This allows them to easily permeate every cell, both healthy and unhealthy. The CPPs lack cell-type specificity, which hampers their in vivo application.
Many innovative strategies have been developed over time to enhance the selectivity of the CPPs for the cancerous cells. One promising technique involves the use of peptidomimetics- artificially developed peptide-like compounds designed to enhance specificity, stability and therapeutic performance.
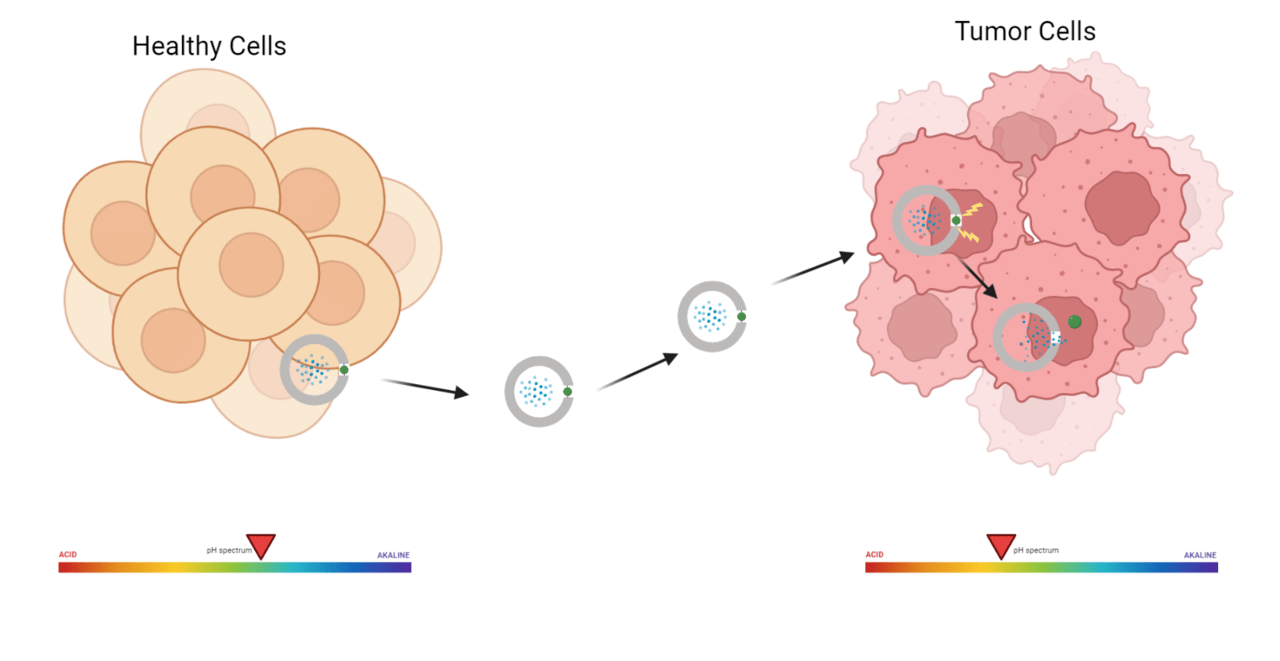
This work discusses the development of a new peptidomimetic (Hkd) that can selectively target cancerous cells, enter them, and deliver an anticancer drug. The ability of this peptidomimetic to differentiate between cancer cells and the non-cancerous ones is understood from its design. The structure of the peptidomimetic, (Hkd) a decamer, comprises histidine and a CDP amino acid (cyclo-Lys-Asp). This confers pH sensitivity to this new and unconventional peptide, as unlike the previously developed peptide stretches, which were Lysine- and arginine-rich, this peptide contains histidine. The imidazole group of histidine does not easily pick up a proton under normal biological conditions, because it requires relatively acidic environments to become protonated. This ensures that the peptide Hkd doesn’t penetrate normal tissues and only enters those that offer an acidic environment. It requires an acidic environment to protonate the imidazole nitrogen, making it positively charged and thereby cell-permeable. This is a significantly better option compared to conventionally used CPPs, which are typically rich in lysine and arginine. Lysine has a higher pKa and hence is always protonated. The permanent positive charge on the body of these peptides facilitates smooth and indiscriminate cell penetration (that is, they easily enter both cancerous and non-cancerous cells). Another advantage of the newly developed peptide’s structure is the unconventional CDP amino acid, cyclo (Lys-Asp). This amino acid confers resistance to proteolytic enzymes, thereby enhancing the peptide’s stability in the bloodstream. The cyclic structure of this residue also makes it more rigid. In addition, the CDP core offers multiple hydrogen bonding donor and acceptor sites, which facilitate selective interaction, mediate cellular uptake, and enhance biocompatibility.
The decamer Hkd has the unnatural amino acid CDP (kd) and natural amino acid histidine (H) present at alternate positions, each conferring the functions discussed above. Histidine’s pH-dependent protonation helps selectively target cancer cells, while the hydrogen bonding interactions between CDP and the cell membrane facilitate the internalization of the decamer. This peptide was tethered to an anti-cancer drug, Camptothecin (Cpt) for its application.
To understand and determine the role of CDP in evading enzymatic degradation, stability studies were conducted for both Hkd, the peptidomimetic, and Cpt-Hkd, the Camptothecin-peptidomimetic conjugate. Tests conducted in human blood serum (HBS) were analyzed using HPLC. It was observed that after 48 hours of incubation in HBS, more than 75% of the peptidomimetic remained integrated. Very similar stability was observed for the drug-peptidomimetic (Cpt-Hkd) conjugate. These tests indicate the extra stability imparted by CDP to the entire scaffold.
Cytocompatibility of the peptidomimetic, Hkd, was monitored in HeLa (Human Cervical Cancer cell line) and L929 (non-cancerous murine cell line) cells. It was observed that the cell viability remained greater than 90% across the varying concentrations of Hkd tested. This demonstrates the biocompatibility of the synthesized peptidomimetic and confirms its suitability for in vitro application. In cancer therapy, biocompatibility matters because delivery systems such as peptidomimetics must be safe and well tolerated by the body, even when they are used to ferry highly toxic chemotherapeutic agents to tumors.
Following this, pH-dependent cellular uptake studies were conducted. The fluorophore fluorescein isothiocyanate (FITC) was tethered to Hkd and Cpt-Hkd to monitor their behaviour using flow cytometry. Hkd, as anticipated, showed a weak
membrane permeability in HeLa cells at physiological pH. However, upon lowering the pH of the medium to 6.8 and 6.0, cellular uptake of Hkd gradually increased, by 1.8-fold and 3.2-fold, respectively, compared to pH 7.4.
The in vitro cytotoxicity of the Cpt-Hkd conjugate was evaluated in L929 cells (a non-cancerous murine cell line). The free drug, Cpt, showed significant toxicity towards L929 cells, with the cell viability going down to 30% at just 20µM drug concentration. The non-specific interactions and greater toxicity of the free drug are attributed to its hydrophobic core structure, which facilitates membrane penetration. The toxicity of Cpt towards normal cells decreased significantly when conjugated with the peptidomimetic (Hkd). The Cpt-Hkd conjugate was found to be less toxic to cells, with cell viability increasing by 63%.
In conclusion, the scientists developed a peptidomimetic that could take advantage of the slightly acidic condition, a characteristic feature of tumours. By bringing together a rigid molecular scaffold with a pH-responsive amino acid, the molecule could efficiently pass through the cell membrane and remain stable long enough to be able to discharge its function. Importantly it preferentially targeted cancer cells over normal cells. When conjugated with an anti-cancer drug camptothecin, it selectively entered and destroyed cancer cells in an acidic environment and greatly lowered unwanted toxicity in normal cells.
This shows how careful construction of smart drug carriers can improve drug delivery, reduce side effects and help overcome drug resistance in cancer treatment.
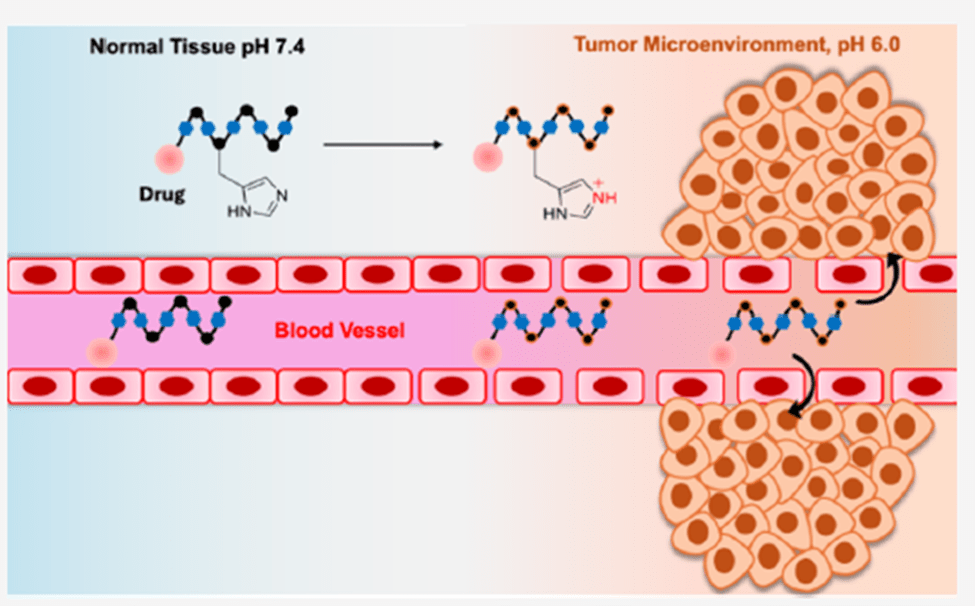
Header Image Caption & Source: Illustration of a gated drug delivery system retaining its cargo until it encounters the acidic tumor environment. Wikimedia Commons
In-text Image Caption & Source: Illustration of the HkD peptidomimetic continuing to carry an anticancer drug until it encounters the acidic tumor environment and its histidine residue gets protonated. Picture courtesy- Biochemistry 2025, 64, 1266−1275.
Edited by Karli Norville
References
[1] B. Maity, H. Moorthy, T. Govindaraju, Biochemistry 2025, 64, 1266-1275.

{kind=link}
Leave a comment